Far beyond the wavelength limit
This system in the old AS building is an optical microscope, but is
a far cry from the classic combination of objective, ocular, and focusing screw on a curved frame. Here the objective is below the specimen holder, and the ocular has been replaced by a super-sensitive and very fast black-and-white camera. The technique for seeing details smaller than the wavelength of light was first used in 2006. According to classic physics, this is impossible, but the technique of single molecule localisation microscopy (SMLM) actually transforms a microscope into a nano-scope. In 2014, the inventors of this new technique (Eric Betzig, Stefan Hell and William Moerner) were awarded the Nobel Prize in chemistry for their work. At TU Delft, Prof. Bernd Rieger and Prof. Sjoerd Stallinga (Faculty of Applied Sciences, AS) work with this technique to make biomolecules visible. Their mission is to reveal increasing levels of detail of biomolecules in their natural surroundings. In doing so, they are pushing the envelope of what is physically and technically possible. Although classic light-based microscopy can only reveal details down to 250 nm, details can be visualised using SMLM down to approximately 10-20 nm, and researchers at TU Delft are aiming for ‘a few nanometres’.
Target molecule
In their structural studies of cellular nuclear pores, they added fluorescent molecules (fluorophores) that bind to the inner and outer edge of the target molecule complex. A cell nucleus can easily have a thousand pores, all of which can emit light. The exposure time lasts approximately an hour, and there are also hundreds of images of cellular nuclear pores in all possible orientations. Using this data, Rieger and Stallinga calculate the structure of the target molecule. This computation of data fusion or particle averaging requires several hours of computation time, resulting in a 3D image of a cellular nuclear pore with details down to the level of a nanometre (59 nm deep and 96 nm wide).
DNA twist
When a fluorophore emits light, the light is not distributed evenly in all directions. The spatial distribution resembles a halter more than a sphere. PhD candidates working with Stallinga and Rieger realised that this ‘anistropy’ makes it possible to deduce the orientation of a molecule from the light intensity. They then colour-coded the orientations, thereby displaying the twisting of two DNA strands as repetitive colour patterns for the first time using light. Last spring (2022), Stallinga was awarded a European research grant (ERC advanced grant) for his proposal to utilise scanning with non-uniform lighting. In doing so, he hopes to make super-resolution microscopy even more precise while at the same time analysing a larger volume. It’s no less than astonishing to see that, 350 years after Van Leeuwenhoek, the optical microscope is still being further improved.

©Sam Rentmeester
Bernd Rieger and Sjoerd Stallinga make biomolecules visible.

It was the first time that DNA twisting was visualised using light
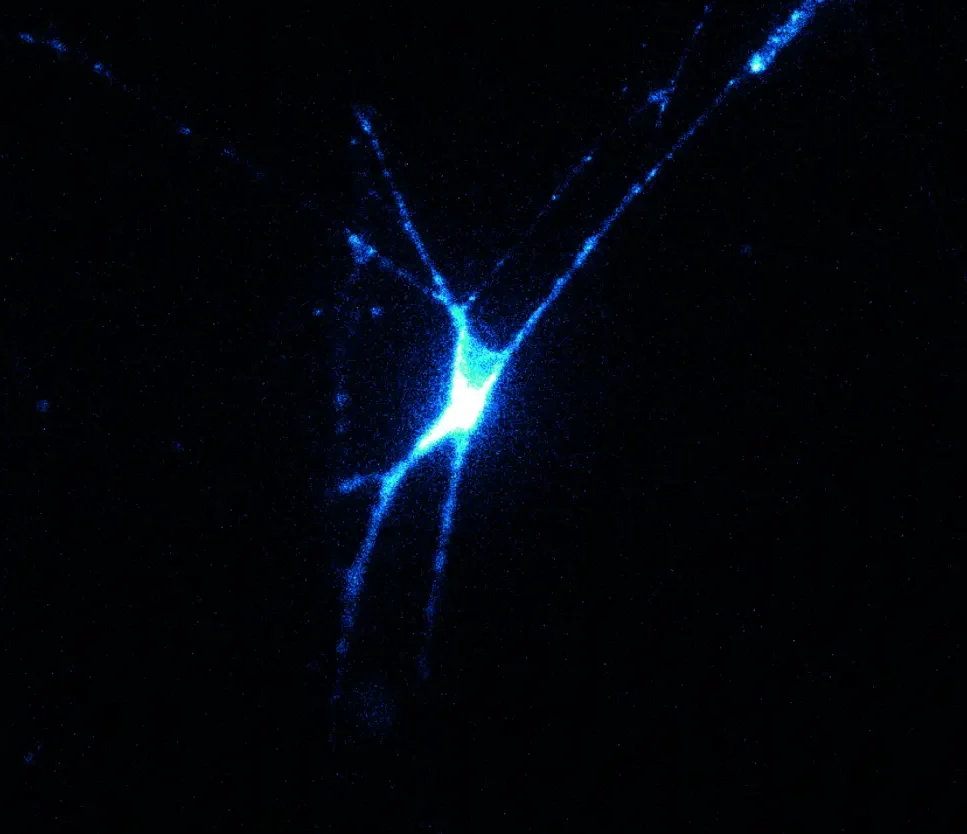
©Sam Rentmeester
Microscope image of a nerve cell with a fluorescent voltage sensor.
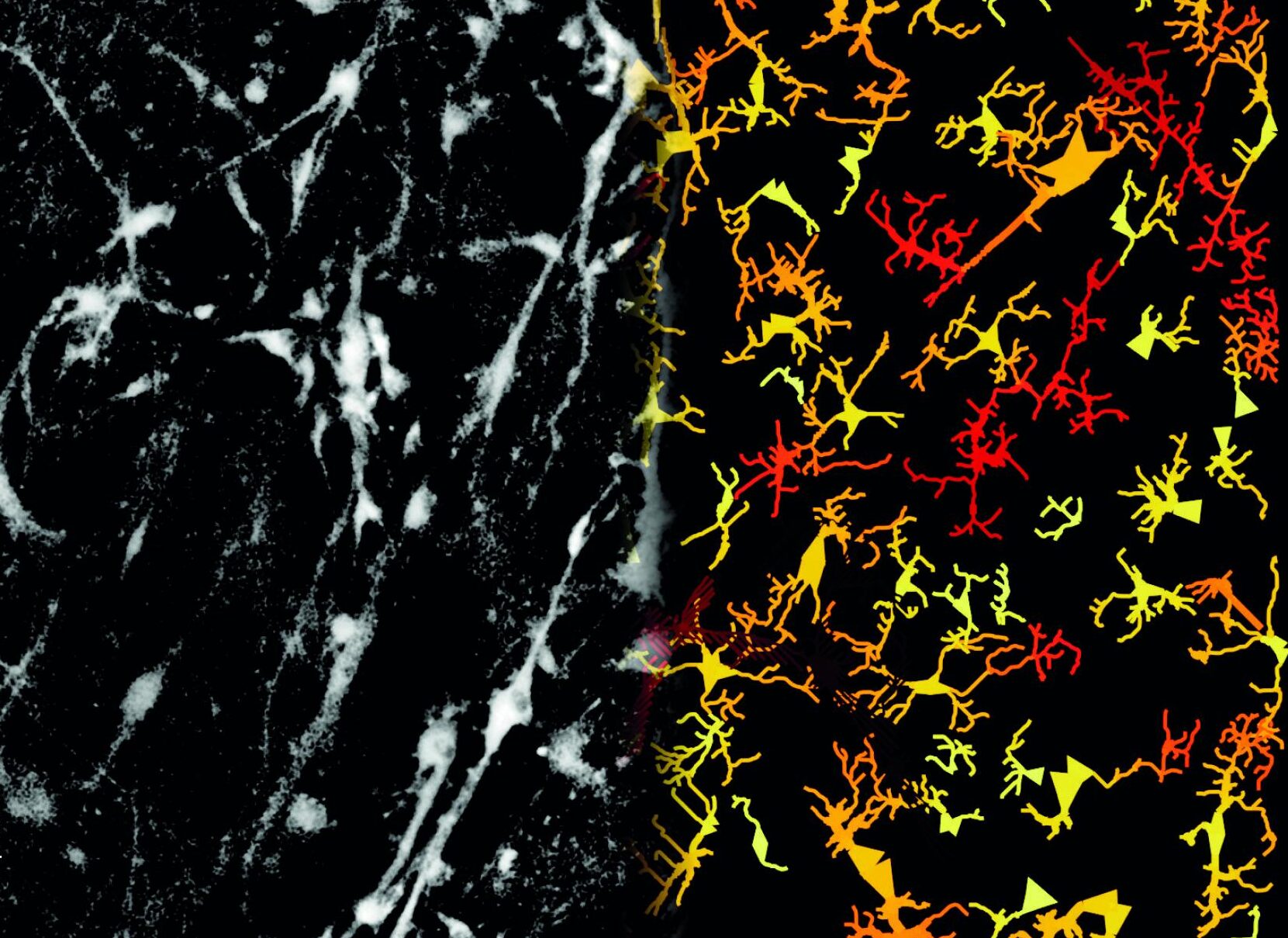
© Tijn Hoekstra
Microscoopbeeld van een neuraal netwerk (links) en de daaruit afgeleide computationele structuur (rechts).
Onderzoekers kunnen met deze techniek in het levende brein van een muis kijken

© Sam Rentmeester
Microscope image of a neural network (left) and the derived computational structure (right).
Researchers use this technique to look inside the living brain of mice

© Sam Rentmeester
Arjan Jakobi is specialized in cryo-EM, whereby the narrow bundle of electrons is focused on a deep-frozen (cryogenic) protein sample.
Seeing the brain at work
It was a striking video shown by Dr Daan Brinks. He heads the Brinks Lab with laboratories at TU Delft and the Erasmus MC in Rotterdam. The slogan of his lab is ‘imaging the brain at work’ and that is exactly what the video shows: flashes of light travelling through a network of nerve cells. But this is no animation. The slogan is not about ‘imagining’ but ‘imaging’. The video is a slow-motion optical microscope registration of pulses travelling through a living neural network. Using ‘voltage imaging’ the Brinks Lab is not making the structure of neural networks visible but rather their activity. How do they do this? The electrical pulses are caused by very brief laser flashes lasting about 1 ms. This is done using a technique called ‘optogenetics’, which is so smart and versatile that both Nature and Science called it the ‘method of the year’ (respectively ‘of the decade’) in 2010. As nerve cells contain a light-sensitive ion channel, a laser flash triggers an action potential. As soon as this molecular complex is bombarded by a flash of light, ions flow inwards and cause an action potential, which then travels across neural branches to neighbouring neurons. Cells do not naturally have light-sensitive gateways, but the cell builds one after a viral infection with synthetic DNA. “We introduce the synthetic DNA through a virus,” explains Brinks, “allowing us to change specific cells.”
Molecular voltage meters
However, this doesn’t explain how changes in potential are actually made visible. The Brinks Lab developed molecular voltage meters to do this: fluorescent proteins (archaerhodopsin proteins) that emit more light as the electrical voltage over the molecule increases. The concept of molecular voltage meters is already several decades old, but the first working versions were recently developed in Adam Cohen’s lab at Harvard, where Brinks did his postdoc. Brinks’ lab at TU Delft is now developing smart microscopes and improved molecular voltage meters to adapt the technology for use in neuroscientific research and clinical diagnostics.
At the Erasmus MC, researchers use this technology to look inside the living brain of a mouse. They try to visualise changes in the pattern of light flashes when the mouse learns new tricks such as licking something or blinking its eyes on command. Brinks calls this the ‘biophysical manifestation of the learning process’. Brinks is searching for the ‘physical basis of the brain’. Supplementing psychology and anatomy, his lab is trying to find out what happens at the physical cellular level. As an example, he mentions the study of nerve cells from an ALS patient, grown from stem cells. As opposed to healthy nerve cells, ALS neurons do not produce a stronger signal when the sensory stimulus increases but rather a weaker one, falling silent. The question is why?!
Freeze! Proteins in a bundle of electrons
Proteins are the multifunctional tools of living cells: they build
and repair bodily tissues, facilitate biochemical reactions and communication, and give structure to cells, to name just a few of their functions. They are made of long chains of 20 different amino acids, and what they do depends on their structure. That structure is determined by how the protein is folded in an aqueous environment: hydrophobic elements stick together and form the core, while hydrophilic elements compose the outside. Researchers would very much like to know the precise 3D structure of proteins. That knowledge allows pharma companies, for example, to design a small molecule that can block a specific protein and stop undesirable processes (e.g. infection). The most powerful microscope is the electron microscope, but this, including the vacuum chamber and bundle of electrons, is disastrous for proteins. That, in short, is the challenge faced by a special branch of microscopy, namely Cryo-EM – the specialty of Dr Arjen Jakobi (Department of Bio-nanoscience at the Kavli Institute of Neuroscience at AS). In Cryo-EM, the narrow bundle of electrons is focused on a deep-frozen (cryogenic) protein sample. This is not as simple as it sounds. Two years ago, Jakobi was awarded a European research grant (ERC starting grant) to build his group of researchers. This year, another ERC grant will be awarded to his group to develop a cryo-chip to make the imaging process more efficient.
Ultra-fast freezing process
“A protein is a sensitive molecule that retains its natural form only
in aqueous environments,” explains Jakobi. “But the vacuum causes any water present in the electron microscope to immediately evaporate, besides which the samples also have to be ‘fixed’.” This problem can be solved by freezing the proteins dissolved in liquid. This has to be done at lightning speed (from 20° to -200°C in 1 ms) to avoid ice crystals forming that would distort the proteins. For this snap freezing, the group uses a setup that inserts the sample into liquid ethane at very high speed. From there, it moves (immersed in liquid nitrogen) to the electron microscope. Bundle intensity is minimised so as not to damage the proteins. The image obtained is an almost even grey colour, but it still contains shadows of hundreds of thousands of molecules in all possible positions in a very thin layer of ice 20 to 100 nm thick. The computational process used to deduce the
3D structure of the protein from all these tens of thousands of projections is called back projection. After days or even weeks of computation, an unbelievably sharp 3D image of the protein appears where you can almost distinguish the individual atoms. Jakobi is now working on registering the motions of proteins and their interactions with other molecules. “Then we can really understand how a protein can do what it does.”
On January 13, 2023, Bernd Rieger and Sjoerd Stallinga will talk about their work during the 181st Dies Natalis of TU Delft. You can follow this presentation online.